Getting Started
Torben Kimhofer
2026-05-10
Source:vignettes/getting-started.Rmd
getting-started.RmdIntroduction
This vignette demonstrates data import and preprocessing workflow for NMR-based metabolomics with metabom8.
Importing spectral data
In routine high-throughput NMR workflows, raw time-domain data (the free-induction decay) are typically processed directly on the spectrometer using the vendor software TopSpin. Downstream metabolomics analysis therefore commonly starts from frequency-domain spectra. Consequently, this vignette focuses on importing spectra generated by TopSpin.
For completeness and teaching purposes, the section (Starting point: raw FID) briefly illustrates importing and transforming time domain data to obtain spectra.
TopSpin-processed spectra
The function read1d_proc() imports TopSpin-processed 1D
NMR spectra from the pdata subdirectory of Bruker
experiment folders (typically pdata/1). It reads the
processed absorption-mode spectrum (e.g. 1r) together with
associated acquisition (acqus) and processing
(procs) parameters.
At minimum two arguments are required:
-
path: path to the directory that encloses NMR experiments -
exp_type: list of spectrometer parameters to filter desired experiment types based on acquisition and processing metadata.
Further details on supported filtering options are provided in
?read1d_proc.
## Example: import 1D spectra
exp_dir <- system.file("extdata", package = "metabom8")
exp_type <- list(pulprog="noesygppr1d")
nmr_data <- read1d_proc(exp_dir, exp_type, n_max = 100)
#> Imported 2 spectra.
names(nmr_data)
#> [1] "X" "ppm" "meta"Both import functions (read1d_proc() and
read1d_raw()) return a named list containing three core
components used throughout this vignette: the spectral matrix
(X), the chemical-shift axis (ppm), and
associated metadata (meta).
For interactive exploration, setting to_global = TRUE
assigns the components X, ppm, and
meta directly to the global environment
(.GlobalEnv). Existing objects with these names will be
overwritten.
The row names of X and meta correspond to
experiment directories and can be used to join external sample
annotations. The meta data frame contains full TopSpin
acquisition and processing parameters for each spectrum.
Example acquisition and processing parameters as they appear in
meta:
| Prefix | Meaning | Examples |
|---|---|---|
| a_ | Acquisition parameters | a_PULPROG, a_SFO1, a_RG, a_SW_h, a_TE |
| p_ | Processing parameters | p_WDW, p_LB, p_SI, p_PHC, p_BC, p_NC_proc |
See ?read1d_proc() for further details.
Raw FID data
For completeness, metabom8 also provides functionality
to import and process raw FIDs. This allows users to reproduce basic
spectral processing steps within R or to explore how different
processing parameters influence the resulting spectra.
Processing an FID converts the time-domain signal into a frequency-domain spectrum. Typical processing steps include digital-filter correction (group-delay), apodisation (windowing), zero-filling, Fourier transformation, phase correction.
The function read1d_raw() performs these operations:
# import 1D NMR data
nmr_raw <- read1d_raw(
exp_dir,
exp_type,
apodisation = list(fun = "exponential", lb = 0.2),
zerofil = 1L,
mode = "absorption"
)
#> Processing 2 experiments.
names(nmr_raw)
#> [1] "X" "ppm" "meta"The following examples illustrate several commonly used apodisation
profiles implemented internally in metabom8.
# selected FID apodisation functions
f_apod <- c("sine","cosine","exponential","sem")
# create toy fid
n <- 200; t <- seq(0,1,len=n)
fid <- exp(-5*t)*cos(20*pi*t)
# apply apodisation
A <- sapply(f_apod, \(f) metabom8:::.fidApodisationFct(n, list(fun=f, lb=-0.2)))
Fp <- sweep(A, 1, fid, `*`)
S <- apply(Fp, 2, \(x) Mod(fft(x))[1:(n/2)])
# compare graphically
cols <- 1:ncol(A)
par(mfrow=c(2,2), mar=c(3,3,2,1))
plot(t, fid, type="l", lwd=2, main="FID")
matplot(t, A, type="l", lwd=2, col=cols, main="Apodisation windows")
matplot(t, Fp, type="l", lwd=2, col=cols, main="Windowed FID")
matplot(S, type="l", lwd=2, col=cols, main="Spectra")
legend("topright", f_apod, col=cols, lty=1, bty="n", cex=.8)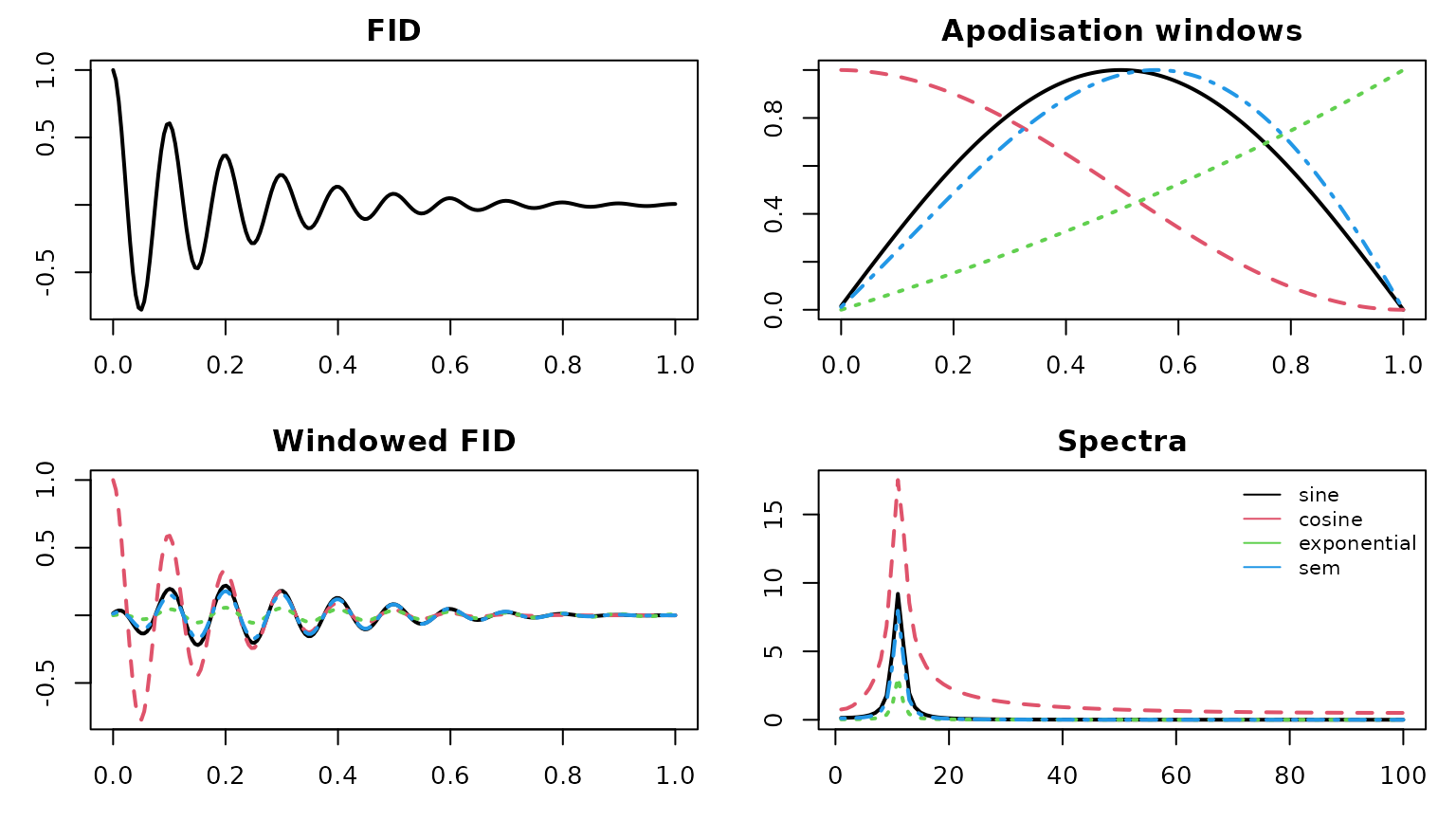
Visual inspection
Spectra can be visualised with the function plot_spec().
The minimum set of arguments include the spectral data X
(array or matrix) and the chemical shift vector ppm.
Three plotting backends are available: "plotly"
(default), "base", and "ggplot2". The
"plotly" backend is set up to use WebGL, enabling smooth
interactive graphics even when the number of data spectra is large. The
"ggplot2" backend is not iteractive and renders much
slower. However, the returned object integrates with the extensive
"ggplot2" ecosystem and can be further customised to
generate publication-quality figures.
See ?plot_spec for additional details.
data("covid", package = "metabom8")
## plot directly from the dataset object
plot_spec(covid, shift = c(0.5, 2))
## alternatively extract data components: X, ppm
X <- covid$X
ppm <- covid$ppm
dim(X)
#> [1] 10 27819
head(ppm)
#> [1] 4.549818 4.549513 4.549207 4.548902 4.548596 4.548291
plot_spec(X[1:3, ], ppm, shift = c(0.5, 2)) # backend='plotly' by default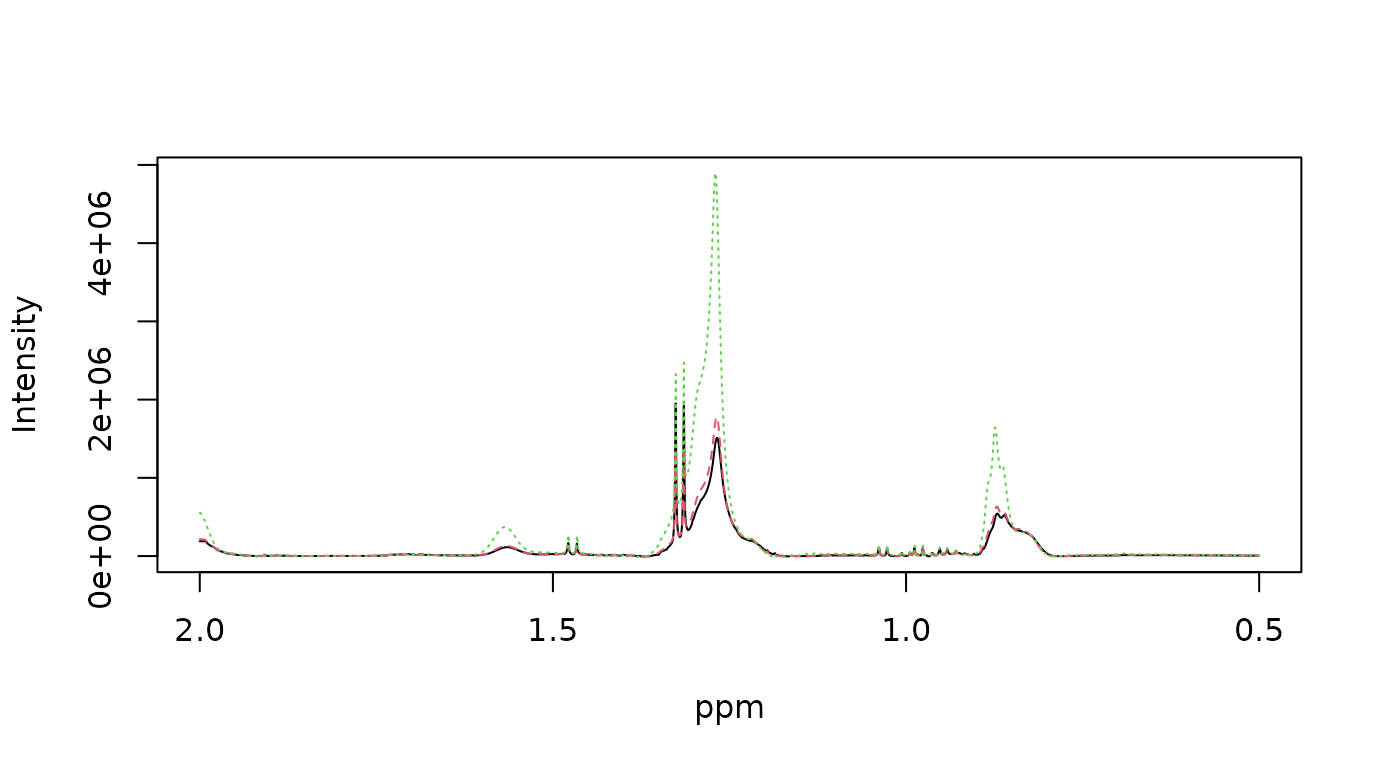
Preprocessing
metabom8 provides a modular preprocessing API for 1D NMR
spectra. Each preprocessing function is composable and designed to
support both interactive exploration and reproducible pipelines.
For a list of preprocessing functions call
list_preprocessing() from the R console.
Pipeline-style usage
When operating on a metabom8-style dataset object,
preprocessing functions return the same structured object: a named list
containing the elements X, ppm, and
meta. This allows steps to be chained using the base R pipe
operator (|>):
data("hiit_raw", package = "metabom8") # urine NMR (HIIT exercise experiment)
names(hiit_raw) # X, ppm, meta
#> [1] "X" "ppm" "meta"
# piped preprocessing
hiit_proc <- hiit_raw |>
calibrate(type = "tsp") |>
excise() |>
correct_baseline(method='asls') |>
align_spectra() |>
pqn()
dim(hiit_proc$X)
#> [1] 3 54009This API design enables clear, declarative workflows suitable for production analyses.
Stepwise execution
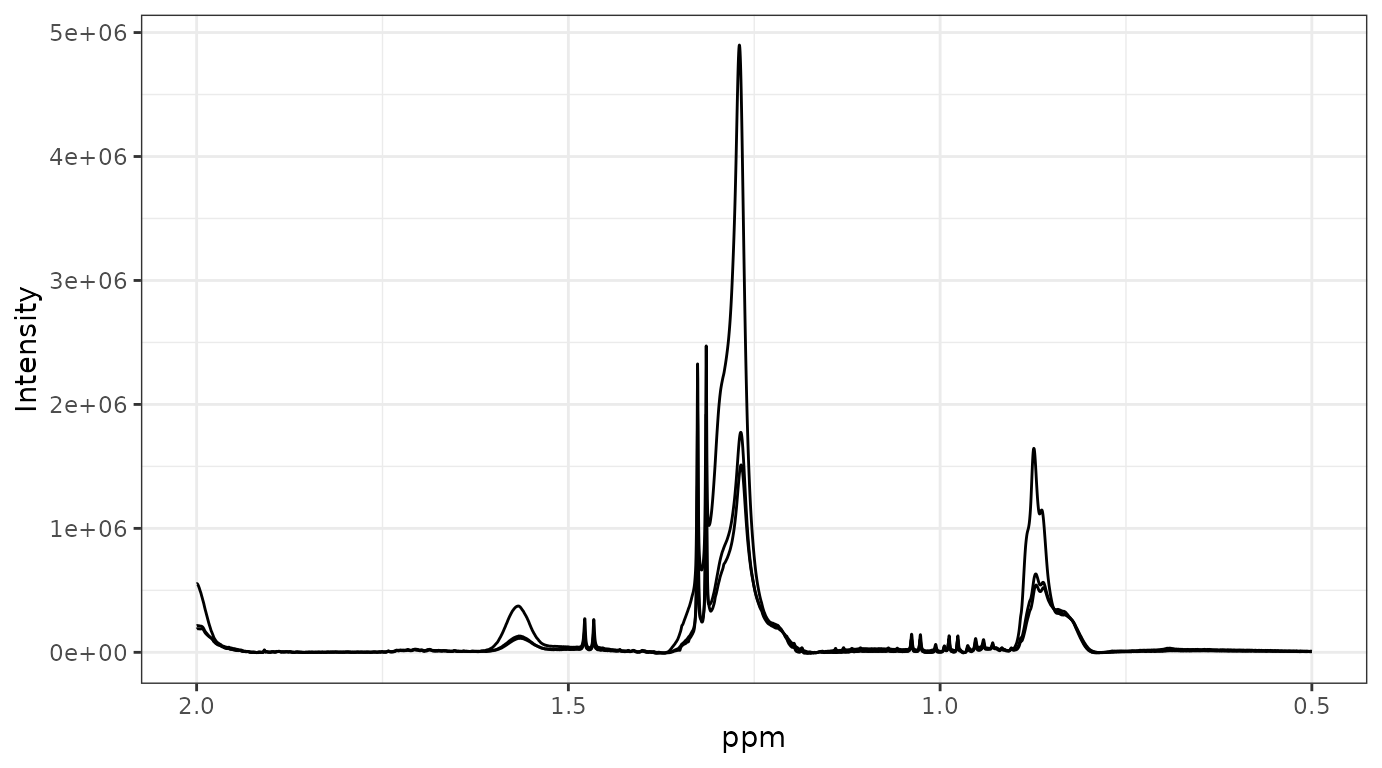
Each operation can also be applied independently to a spectral matrix and ppm vector. This explicit alternative is useful for pipeline development, parameter tuning and visual inspection.
X <- hiit_raw$X
ppm <- hiit_raw$ppm
# perform TSP calibration
X_cal <- calibrate(X, ppm, type='tsp')
# excise chemical shift regions
regions <- list(
upfield_noise = c(min(ppm), 0.25),
residual_water = c(4.5, 5.2),
downfield_noise = c(9.7, max(ppm))
)
X_exi <- excise(X_cal, ppm, regions)
ppm_exi <- attr(X_exi, 'm8_axis')$ppm
# baseline correction
X_bli <- correct_baseline(X_exi)
# plot spectrum
plot_spec(X_bli[1,], ppm_exi, shift = c(0,10))Provenance
Data provenance refers to the documented record of how data were generated, processed, and transformed. Recording this information ensures that analyses remain reproducible and that individual preprocessing steps can be inspected, verified, or repeated at a later stage.
In metabom8, particular emphasis is placed on
transparent preprocessing, as these decisions can substantially
influence downstream statistical analyses and biological interpretation.
Each preprocessing operation automatically records its parameters and
execution context as structured metadata attached to the spectral matrix
X.
The resulting dataset therefore remains self-describing: the complete processing history is linked to the data matrix and can be inspected programmatically at any time.
## Inspect preprocessing steps
print_provenance(hiit_proc)
#> metabom8 processing pipeline:
#> ==============================
#> [1] calibrate
#> params:
#> target :
#> mode : singlet
#> window : [-0.2, 0.2]
#> centre : 0
#> notes: Chemical shift calibration applied.
#>
#> [2] excise1d
#> params:
#> regions :
#> upfield_noise : [-5.21794, 0.25]
#> water : [4.5, 5.2]
#> urea : [5.5, 6]
#> downfield_noise : [9.7, 14.8037]
#> notes: Specified chemical shift regions removed using get_idx().
#>
#> [3] baseline
#> params:
#> method : asls
#> args :
#> notes: Baseline correction applied.
#>
#> [4] align spectra
#> params:
#> half_win_ppm : 0.007
#> n_intervals : 86
#> lag.max : 200
#> intervals_ppm : <86 items> [[9.68427, 9.66121], [9.46736, 9.44216], [9.29139, 9.27764], ...]
#> notes: Median-derived chemical shift intervals aligned independently using cross-correlation.
#>
#> [5] pqn
#> params:
#> ref_index :
#> total_area : FALSE
#> bin :
#> notes: Probabilistic quotient normalisation applied.
# Retrieve specific step
get_provenance(hiit_proc, step = 2)
#> $step
#> [1] "excise1d"
#>
#> $params
#> $params$regions
#> $params$regions$upfield_noise
#> [1] -5.217943 0.250000
#>
#> $params$regions$water
#> [1] 4.5 5.2
#>
#> $params$regions$urea
#> [1] 5.5 6.0
#>
#> $params$regions$downfield_noise
#> [1] 9.70000 14.80367
#>
#>
#>
#> $notes
#> [1] "Specified chemical shift regions removed using get_idx()."
#>
#> $time
#> [1] "2026-05-10 12:57:01.113241"
#>
#> $pkg
#> [1] "metabom8"
#>
#> $pkg_version
#> [1] "1.1.2"
## Retrieve the full provenance log
log <- get_provenance(hiit_proc)
# print(log) # very detailedEach entry stores:
- sequential execution index
- preprocessing step name
- parameter settings
- optional notes
- timestamp and versioning info
Individual parameter values can be retrieved as well, allowing the full transformation history of a dataset to be inspected programmatically.
# Retrieve parameter from a named step
get_provenance(hiit_proc, step = "calibrate", param = "target")
#> $mode
#> [1] "singlet"
#>
#> $window
#> [1] -0.2 0.2
#>
#> $centre
#> [1] 0Custom notes can also be included in the provenance log. This is particularly userful when data are processed in automated fashion, for example when using workflow managers:
# collect env varaibles
params <- list(
agent = paste0("snakemake/", Sys.getenv("SNAKEMAKE_VERSION", "v?")),
runtime = "docker",
workflow = Sys.getenv("M8_WORKFLOW", "std_prof-urine"),
run_id = Sys.getenv("M8_RUN_ID", "m8-2605-001")
)
# create note with env parameters
hiit_proc <- add_note(hiit_proc,
'Excluding outlier sample XX - reason is ...',
params)
# check provenance out
get_provenance(hiit_proc , step = "user note")
#> $step
#> [1] "user note"
#>
#> $params
#> $params$agent
#> [1] "snakemake/v?"
#>
#> $params$runtime
#> [1] "docker"
#>
#> $params$workflow
#> [1] "std_prof-urine"
#>
#> $params$run_id
#> [1] "m8-2605-001"
#>
#>
#> $notes
#> [1] "Excluding outlier sample XX - reason is ..."
#>
#> $time
#> [1] "2026-05-10 12:57:09.20693"
#>
#> $pkg
#> [1] "metabom8"
#>
#> $pkg_version
#> [1] "1.1.2"Note on provenance
metabom8records preprocessing history as structured attributes on standard R matrices or named lists. This design is lightweigth and prioritises interoperability and flexibility over strict container classes (e.g.,SummarizedExperiment). Provenance is preserved when objects are saved/loaded based on standard R serialisation (save()/saveRDS(),load()), but may be lost if the object is modified by functions outside themetabom8API that drop or overwrite attributes (for example when objects are coerced, reconstructed, or exported to external formats).
Modelling
The section demonstrates data modelling using PCA (unsupervised) and PLS/OPLS (supervised).
All modelling calls return an instance of class
m8_model, which can be used for further processing and
diagnostics.
Y <- covid$an$type
X <- covid$XTo prevent the natural dynamic range of metabolite concentrations from dominating statistical modelling, data are scaled prior to analysis. In metabom8, scaling and centering are passed explicitly to the model function:
# Define the scaling and centering strategy
uv = uv_scaling(center = TRUE)See ?uv_scaling for further details and additional
scaling strategies.
Principal Components Analysis (PCA)
Unsupervised modelling with PCA is done by an iterative procedure (NIPALS) that calculates components iteratively, one at a time. Therefore, the number of desired components is provided in the function call.
Here we compute two principal components using unit-variance scaling:
pca_model <- pca(X, scaling = uv, ncomp=2)
# show / summary methods
show(pca_model)
#>
#> m8_model <pca>
#> ----------------------------------------
#> Dimensions : 10 samples x 27819 variables
#> Mode : unsupervised
#> Preprocess : center | UV
#> Components : 2
#> ----------------------------------------
#> Use summary() for performance metrics.
summary(pca_model)
#> $perf
#> comp R2X R2X_cum selected fit
#> 1 1 0.3023058 0.3023058 TRUE fitted
#> 2 2 0.1898503 0.4921561 TRUE fitted
#>
#> $engine
#> [1] "pca (nipals)"
#>
#> $y_type
#> [1] "unsupervised"Partial-Least Squares (PLS)
Partial-Least-Squares (PLS) as supervised modelling technique utilises a statistical re-sampling techniques to determine the optimal number of components and to safeguard against overfitting.
In this vignette, we employ balanced Monte Carlo cross-validation
(balanced_mc). This process repeatedly splits the data into
training and testing sets to ensure the model predictive power
generalizes well to unseen data.
mc_cv <- balanced_mc(k=15, split=2/3, type="DA")See ?balanced_mc for further details and additional
statistical validation strategies.
pls_model <- pls(X, Y, validation_strategy=mc_cv, scaling = uv)
#> Performing discriminant analysis.
#> A PLS-DA model with 1 component was fitted.
# model information
show(pls_model)
#>
#> m8_model <pls>
#> ----------------------------------------
#> Dimensions : 10 samples x 27819 variables
#> Mode : classification
#> Preprocess : center | UV
#> Components : 1 (2 tested)
#> Validation : BalancedMonteCarlo (k = 15)
#> Stop rule : cv_improvement_negligible
#> ----------------------------------------
#> Use summary() for performance metrics.
# model performance summary
summary(pls_model)
#> $perf
#> comp AUCt AUCv R2X selected fit stop_reason
#> 1 1 1 1 0.2502977 TRUE fitted <NA>
#> 2 2 1 1 0.2179150 FALSE not fitted cv_improvement_negligible
#> stop_metric stop_delta
#> 1 NA NA
#> 2 1 0
#>
#> $engine
#> [1] "pls"
#>
#> $y_type
#> [1] "DA"
# model scores & loadings
Tp <- scores(pls_model)
Pp <- loadings(pls_model)Orthogonal-Partial-Least Squares (OPLS)
OPLS can be though of as PLS with an integrated orthogonality filter, which has been reported to improve model interpretability. While standard PLS mixes predictive and non-predictive variation, OPLS partitions the variance into two parts: variation correlated to the response () and variation “orthogonal” (unrelated) to it.
Similar to PLS, the OPLS algorithm evaluates model performance using statistical re-sampling techniques to ensure the results are robust and not driven by noise.
opls_model <- opls(X, Y, validation_strategy=mc_cv, scaling = uv)
#> Performing discriminant analysis.
#> An O-PLS-DA model with 1 predictive and 1 orthogonal components was fitted.
# model information & performance
show(opls_model)
#>
#> m8_model <opls>
#> ----------------------------------------
#> Dimensions : 10 samples x 27819 variables
#> Mode : classification
#> Preprocess : center | UV
#> Components : 2 (3 tested)
#> Validation : BalancedMonteCarlo (k = 15)
#> Stop rule : cv_improvement_negligible
#> ----------------------------------------
#> Use summary() for performance metrics.The OPLS algorithm fitted one predictive and one orthogonal
component. A second orthogonal component was evaluated but the
improvement in cross-validated class prediction (AUCv)based
on the left-out sets was negligible (stop reason).
Therefore, a third component was not fitted to the full spectral
matrix.
summary(opls_model)
#> $perf
#> comp_total comp_pred comp_orth AUCt AUCv R2X selected fit
#> 1 2 1 1 1 1 0.2860832 TRUE fitted
#> 2 3 1 2 1 1 NA FALSE not fitted
#> stop_reason stop_metric stop_delta
#> 1 <NA> NA NA
#> 2 cv_improvement_negligible 1 0
#>
#> $engine
#> [1] "opls"
#>
#> $y_type
#> [1] "DA"For detailed information on component stopping criteria used in
opls() andpls() see
?.evalFit().
Methods implemented for both, pls() and
opls(), models include scores(),
loadings() and fitted() (Y-fitted values from
statistical resampling). For OPLS models, these former two functions
accept argument orth = TRUE to extract orthogonal component
information.
Further OPLS model interpretation can be performed using Variable
Importance in Projection (vip()), which identifies the most
influential variables in the model.
# model scores, loadings & vip
Tp <- scores(opls_model)
To <- scores(opls_model, orth=TRUE)
Pp <- loadings(opls_model)
Po <- loadings(opls_model, orth=TRUE)
vip <- vip(opls_model)To ensure the model is statistically robust, metabom8
provides several diagnostic tools:
-
dmodx(): Calculates the “Distance to the Model X” to identify spectral outliers -
opls_perm(): Executes Y-permutations to validate that the observed is significantly higher than what would be expected by chance. -
cv_anova(): Performs a Cross-Validated ANOVA to test the significance of the model’s predictive ability (only for regression)
# distance to the model in X space
dex <- dmodx(opls_model)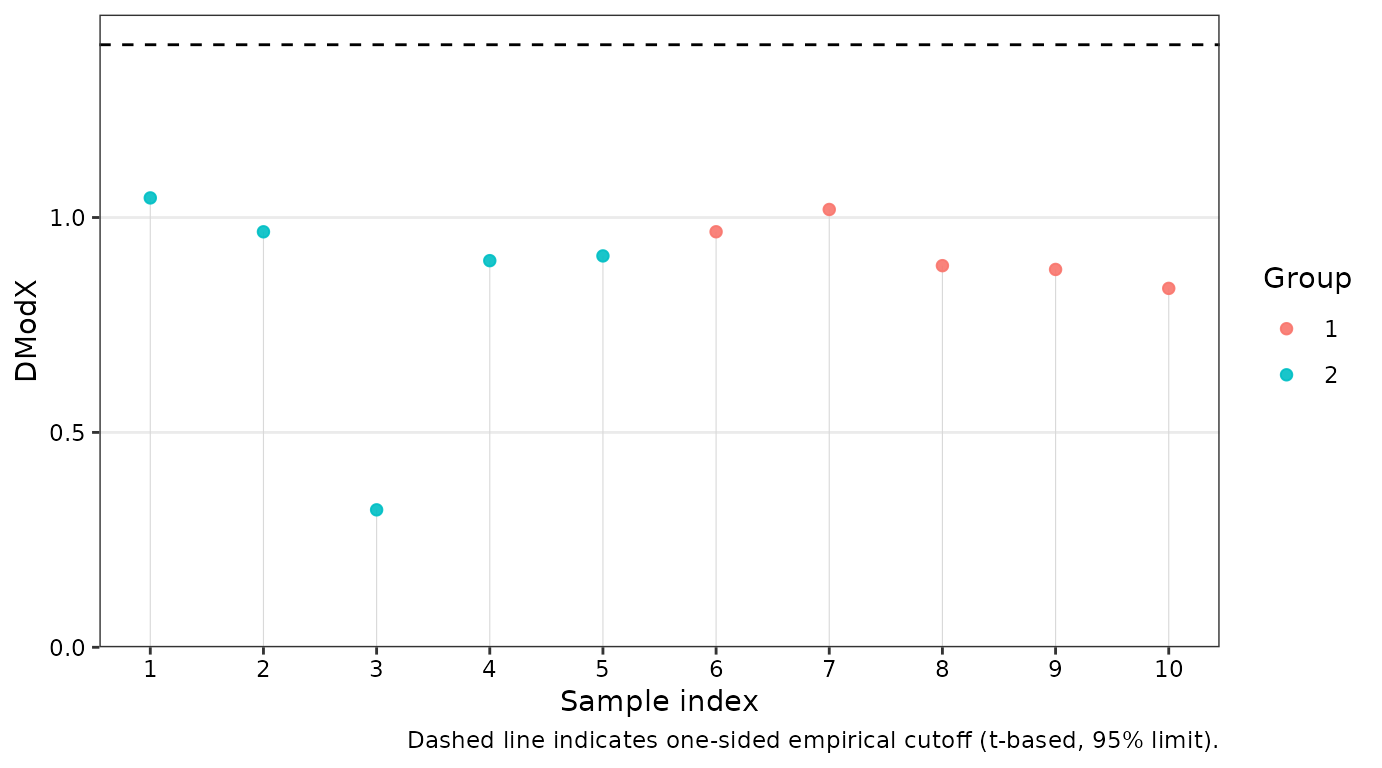
head(dex)
#> ID DmodX passed outlier label Group
#> 1 1 1.0456047 TRUE FALSE 1 2
#> 2 2 0.9667600 TRUE FALSE 2 2
#> 3 3 0.3198258 TRUE FALSE 3 2
#> 4 4 0.8996049 TRUE FALSE 4 2
#> 5 5 0.9106332 TRUE FALSE 5 2
#> 6 6 0.9668600 TRUE FALSE 6 1This vignette demonstrated the core data import, preprocessing, and
modelling workflow implemented in metabom8. For further
details on individual functions, see the corresponding help pages.
Citation
To cite metabom8, please use:
Kimhofer T (2026). metabom8: A High-Performance R Package for Metabolomics Modeling and Analysis. R package version 1.1.2, https://bioconductor.org/packages/metabom8.
A BibTeX entry for LaTeX users is
@Manual{, title = {metabom8: A High-Performance R Package for Metabolomics Modeling and Analysis}, author = {Torben Kimhofer}, year = {2026}, note = {R package version 1.1.2}, url = {https://bioconductor.org/packages/metabom8}, }
Session Info
#> R version 4.6.0 (2026-04-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] metabom8_1.1.2 BiocStyle_2.40.0
#>
#> loaded via a namespace (and not attached):
#> [1] tidyr_1.3.2 plotly_4.12.0 sass_0.4.10
#> [4] generics_0.1.4 stringi_1.8.7 hms_1.1.4
#> [7] digest_0.6.39 magrittr_2.0.5 pROC_1.19.0.1
#> [10] evaluate_1.0.5 grid_4.6.0 RColorBrewer_1.1-3
#> [13] bookdown_0.46 fastmap_1.2.0 plyr_1.8.9
#> [16] jsonlite_2.0.0 progress_1.2.3 BiocManager_1.30.27
#> [19] httr_1.4.8 purrr_1.2.2 crosstalk_1.2.2
#> [22] viridisLite_0.4.3 scales_1.4.0 lazyeval_0.2.3
#> [25] textshaping_1.0.5 jquerylib_0.1.4 abind_1.4-8
#> [28] cli_3.6.6 rlang_1.2.0 crayon_1.5.3
#> [31] ptw_1.9-17 withr_3.0.2 cachem_1.1.0
#> [34] yaml_2.3.12 ellipse_0.5.0 otel_0.2.0
#> [37] colorRamps_2.3.4 tools_4.6.0 parallel_4.6.0
#> [40] reshape2_1.4.5 dplyr_1.2.1 ggplot2_4.0.3
#> [43] RcppDE_0.1.9 vctrs_0.7.3 R6_2.6.1
#> [46] lifecycle_1.0.5 stringr_1.6.0 fs_2.1.0
#> [49] htmlwidgets_1.6.4 MASS_7.3-65 ragg_1.5.2
#> [52] pkgconfig_2.0.3 desc_1.4.3 pkgdown_2.2.0
#> [55] bslib_0.10.0 pillar_1.11.1 gtable_0.3.6
#> [58] data.table_1.18.4 glue_1.8.1 Rcpp_1.1.1-1.1
#> [61] systemfonts_1.3.2 xfun_0.57 tibble_3.3.1
#> [64] tidyselect_1.2.1 knitr_1.51 farver_2.1.2
#> [67] htmltools_0.5.9 labeling_0.4.3 rmarkdown_2.31
#> [70] signal_1.8-1 compiler_4.6.0 prettyunits_1.2.0
#> [73] S7_0.2.2